Neuroendokrine Tumoren (NET) sind eine seltene Form von Krebs mit gerade einmal fünf bis sechs Neuerkrankungen pro 100.000 Menschen, die jedes Jahr diagnostiziert werden.* NET haben ihren Ursprung in neuroendokrinen Zellen und entwickeln sich oft langsam über mehrere Jahre. Neuroendokrine Zellen sind sowohl Bestandteil des endokrinen als auch des Nervensystems und sind überall im Körper zu finden. Diese Zellen geben Neurotransmitter, Zellbotenstoffe oder Hormone in das Blut ab, die die Funktionsweise des Körpers steuern. Das bedeutet, dass sie eine Schlüsselrolle bei der Interaktion zwischen dem Nerven- und dem endokrinen System spielen.
NET entstehen häufig im gastroenteropankreatischen Trakt und werden daher gastroenteropankreatische neuroendokrine Tumoren (GEP-NET) genannt. GEP-NET sind im gesamten Verdauungssystem oder den damit zusammenhängenden Körperbereichen zu finden. Obwohl es sich um eine seltene Krankheit handelt, steigt die Zahl der neu diagnostizierten Patient:innen kontinuierlich an.** GEP-NET verlaufen oft asymptomatisch und sind schwer zu diagnostizieren. Daher werden sie oft erst in einem späten Stadium diagnostiziert, wenn sich bereits Metastasen gebildet haben und die Möglichkeiten einer operativen Entfernung begrenzt sind. GEP-NET treten vorwiegend bei Patient:innen im Alter von 50 - 60 Jahren auf, wobei Frauen etwa 2,5-mal häufiger betroffen sind als Männer.* In einigen Fällen können GEP-NET verschiedene Hormone absondern und werden dann funktionelle GEP-NET genannt.
Operationen sind die Hauptbehandlung für NET und stellen häufig die einzig notwendige Therapieform dar. NET können jedoch in andere Körperteile streuen, oder es ist eventuell nicht möglich, den Tumor ausschließlich operativ zu entfernen. Wenn dies der Fall ist, stehen andere Behandlungsoptionen wie Chemotherapie oder zielgerichtete Therapeutika zur Verfügung.
Das Tumorstadium und der Tumorgrad spielen bei der Behandlung von NET eine wichtige Rolle.
Der Begriff Stadium bezieht sich auf den Entwicklungsstatus des Tumors, d.h. ob er gerade erst entstanden ist, ob er fortgeschritten ist oder ob er bereits in andere Bereiche des Körpers gestreut (metastasiert) hat.
Der Tumorgrad gibt Auskunft darüber, wie schnell der Tumor voraussichtlich wachsen und sich ausbreiten wird. Dazu werden anormale Zellen unter dem Mikroskop mit normalen, gesunden
Zellen verglichen. In der Fachsprache wird dies durch den Ki-67-Wert angezeigt. Zellen eines niedriggradigen Tumors (niedriger Ki-67-Index), der auch als „gut differenziert“ bezeichnet wird, wachsen und breiten sich oft langsam aus. Zellen eines hochgradigen „schlecht differenzierten“ Tumors (hoher Ki-67-Index) wachsen hingegen schnell.
Zu den derzeitigen Standardtherapien für die Behandlung von GEP-NET in der Bauchspeicheldrüse und im Dünndarm gehören beispielsweise Somatostatin-Analoga, Chemotherapie, Immuntherapie, lokoregionale Behandlung (z. B. Zytoreduktionschirurgie, Radiofrequenzablation [RFA], auf die Leber zielende intraarterielle Intervention), die gezielte immunsuppressive Everolimus-Therapie, ein
mTOR-Inhibitor, der Teilung und Wachstum von Krebszellen durch Blockierung des Enzyms mTOR beeinflusst, die gezielte Sunitinib-Therapie, ein multizentrischer Tyrosinkinase-Inhibitor oder eine spezielle Form der zielgerichteten Radiopharmazeutischen Therapie (RPT) mit Lutetium-177-DOTATATE (Lutetium-177-Oxodotreotid).*
Zusätzlich zu den Standardtherapien werden in klinischen Studien neue Behandlungsansätze wie die RPT mit n.c.a. Lutetium-177-Edotreotid untersucht, um festzustellen, ob sie sicher und wirksam sind.
* Bryan Oronsky et al., Neoplasia 2017
** Dasari et al., JAMA Oncol 2017
Die Idee, die Strahlentherapie zur Krebsbehandlung einzusetzen, ist mehr als hundert Jahre alt.
Im Jahr 1895 entdeckte Wilhelm Conrad Röntgen die Röntgenstrahlen als eine neue Form der „Strahlung“, die später zur Entwicklung diagnostischer Bildgebungsverfahren führte. Mit der Entdeckung der Strahlung natürlicher Stoffe und Chemikalien durch Henri Becquerel und den grundlegenden Arbeiten von Marie Curie zur Radioaktivität im Jahr 1896 wurde der Weg für den Einsatz radioaktiver Stoffe als medizinische Behandlungsmöglichkeit geebnet. Bald darauf wurden verschiedene Strahlentherapieansätze für ein breites Spektrum von Erkrankungen, oft auch Krebs, untersucht und entwickelt.
Wie kann die Wirkung der Strahlentherapie präzise genutzt werden, um die Tumorzellen gezielt anzugreifen?
Im Gegensatz zur externen Strahlentherapie, bei der die Strahlung von außerhalb des Körpers angewandt wird, wird die RPT durch die intravenöse Infusion eines Radiopharmazeutikums, das die Tumorzellen genau erkennt, in den Körper eingebracht. In den vergangenen Jahrzehnten haben biomedizinische Untersuchungen zu verschiedenen Tumoreigenschaften und tumorbindenden Molekülen zur Weiterentwicklung der Strahlentherapie hin zur Präzisionsonkologie und zur Entwicklung der RPT beigetragen.
Bei der zielgerichteten radiopharmazeutischen Therapie kommen Radiopharmaka zum Einsatz, die aus einem medizinischen Radioisotop, das zur Zerstörung der Tumorzellen durch die Emission einer geringen Strahlungsmenge eingesetzt wird, und einem tumorspezifischen zielgerichteten Molekül bestehen, das nach dem Schlüssel-Schloss-Prinzip an tumorspezifische Rezeptoren bindet, die auf der Oberfläche der Tumorzellen überexprimiert sind. In Kombination mit einem zielgerichteten Molekül und innerhalb eines maximalen Radius von 1,7 mm setzt das medizinische Radioisotop eine kleine Menge Radioaktivität in das Tumorgewebe frei, die darauf abzielt, die Tumorzellen mit minimalen Auswirkungen auf das umliegende gesunde Gewebe zu zerstören. Die hochpräzise Lokalisierung bietet den möglichen Vorteil, dass das gesunde Gewebe um den anvisierten Tumor nur minimal beeinträchtigt wird. Radiopharmazeutika werden wird in einem Labor unter „Guter Herstellungspraxis“ (Good Manufacturing Practice, GMP) nach strikten zertifizierten internationalen Richtlinien hergestellt.
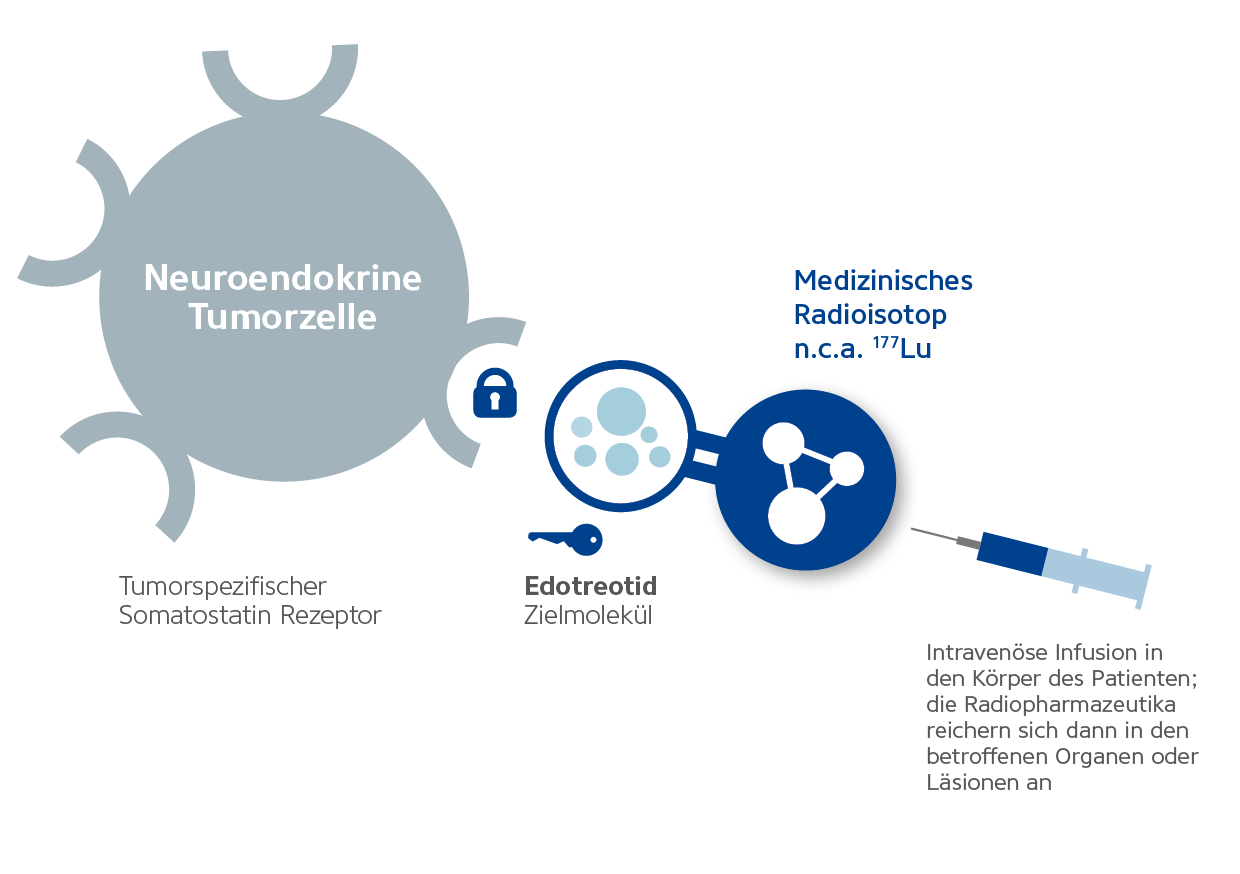
Bei dem Radiopharmazeutikum, das in den klinischen Phase-III-Studien COMPETE und COMPOSE untersucht wird, handelt es sich um n.c.a. Lutetium-177-Edotreotid. Dieses besteht aus zwei Komponenten: dem medizinischen, ungeträgerten (non-carrier-added, n.c.a.) Radioisotop Lutetium-177, das zur Zerstörung der Tumorzellen durch Abgabe einer geringen Strahlungsmenge verwendet wird, und dem zielgerichteten Molekül Edotreotid, einer synthetischen Form des Peptidhormons Somatostatin, das auf neuroendokrine tumorspezifische Rezeptoren abzielt.
Gesunde neuroendokrine Zellen tragen Somatostatin-Rezeptoren auf ihrer Oberfläche. Der Neurotransmitter Somatostatin reguliert das endokrine System innerhalb der normalen Körperfunktionen, indem er nach dem Schlüssel-Schloss-Prinzip an diese Rezeptoren bindet. Die gezielte radiopharmazeutische Therapie nutzt dasselbe Prinzip zur Behandlung von NET. Die Therapie basiert auf der Tatsache, dass neuroendokrine Tumorzellen häufig eine erhöhte Anzahl von Somatostatinrezeptoren auf ihrer Oberfläche exprimieren und damit eine einzigartige Tumoreigenschaft aufweisen, die sich von den Eigenschaften gesunder neuroendokriner Zellen unterscheidet. * Genau wie das natürlich produzierte Hormon Somatostatin bindet Edotreotid an diese Rezeptoren und platziert das medizinische Radioisotop n.c.a. Lutetium 177 direkt auf die erkrankten neuroendokrinen Zellen, sodass es sich an der Tumorstelle anreichert. N.c.a. Lutetium 177 wird in den Tumorzellen internalisiert und zerfällt, wobei medizinische Strahlung (ionisierende β-Strahlung) mit einem maximalen Radius von 1,7 mm freigesetzt und der Tumor zerstört werden. Dank der hochpräzisen Lokalisierung wird das gesunde Gewebe um den anvisierten Tumor herum nur minimal beeinträchtigt.
* Papotti et al., 2002, Virchows Archiv 440(5): 461-75
Bitte beachten: n.c.a. Lutetium-177-Edotreotid ist derzeit in keinem Land zur Vermarktung zugelassen.
Bitte beachten Sie auch die Studieninformationen auf clinicaltrials.gov : COMPETE & COMPOSE
Klinische Prüfungen/klinische Studien spielen eine wichtige Rolle bei der Entwicklung neuer Behandlungsmöglichkeiten. Patient:innen können sich freiwillig für die Teilnahme an solchen Studien entscheiden. Um die Sicherheit der teilnehmenden Patient:innen zu gewährleisten, unterliegen klinische Studien strengen Qualitätskontrollrichtlinien.
Damit eine klinische Studie von einer Gesundheitsbehörde wie den europäischen zuständigen nationalen Zulassungsbehörden oder der U.S. Food and Drug Administration (FDA) genehmigt werden kann, muss die Sicherheit einer neuen Therapie durch eine präklinische Bewertung nachgewiesen werden. Das bedeutet, dass die potenzielle Behandlung in kontrollierter Weise auf mögliche gefährliche Nebenwirkungen getestet wird, bevor sie überhaupt an Patient:innen erprobt wird. Nachdem den nationalen Zulassungsbehörden oder der FDA und den Ethikkommissionen die entsprechenden Informationen vorgelegt wurden, erhält das Medikament die Genehmigung, in die klinische Prüfung zu gehen.
Der Prozess der Arzneimittelentwicklung durchläuft in der Regel vier Phasen, in denen die Wirksamkeit und Sicherheit der Substanz über viele Jahre hinweg an einer wachsenden Zahl von Patient:innen untersucht und beurteilt werden. Wenn das Medikament die klinischen Phasen (I, II und III) erfolgreich durchläuft, wird es in der Regel von der zuständigen Aufsichtsbehörde für die Verwendung in der klinischen Routinepraxis zugelassen.


Sie wünschen weitere Informationen? Nachstehend finden Sie weitere Ressourcen zu den Hintergründen und Verfahren klinischer Studien:
https://www.centerwatch.com/clinical-trials/overview
https://www.clinicaltrials.gov/ct2/about-studies/learn
Weitere Informationen über laufende Studien finden Sie auf den folgenden Webseiten, oder wenden Sie sich an Ihre behandelnde Ärztin/Ihren behandelnden Arzt oder eine Patientenorganisation in Ihrer Nähe.
https://www.clinicaltrials.gov/
https://www.clinicaltrialsregister.eu/